Introduction
Human health is a very sensitive aspect and includes many pathological conditions. Some of these can develop adverse health effects. Thus, inherited diseases pose the greatest danger to the population because they can suppress the health of related individuals over time. Among the multitude of hereditary diseases, special attention should be paid to sickle cell anemia, which manifests itself as a change in the shape of globular proteins. More specifically, in sickle cell anemia, the patient’s standard form of hemoglobin undergoes abnormal mutational changes, resulting in the protein acquiring an unnatural crystalline structure. The pathophysiological manifestations of the disease include multiple edemas and general weakness, which can be regarded as the first signals of the development of the disease. The purpose of this report is to summarize and critically discuss the available scientific information concerning the biochemical side of sickle cell anemia, namely the implementation of mutagenic processes, and metabolic pathways, as well as innovative ways of prevention and treatment.
Structure of Normal Hemoglobin
As is known, human blood is a valuable biological fluid rich in many vital substances, among which hemoglobin is present. Under natural conditions, this molecule is a complex iron-containing tetramer with the ability to bind to oxygen through two metal ion coordination bonds actively (1). Since this type of bond is highly loose and low-strength, the binding of hemoglobin and oxygen leading to the synthesis of oxyhemoglobin is reversible: when this protein enters tissues, oxygen ions are actively sorbed by cells to initiate cellular respiration processes.
The normal form of the hemoglobin molecule is a set of four polyamine acid subunits connected by polypeptide bonds, with each subunit containing a heme: thus, there are up to four hemes per hemoglobin. Hemes are understood as complex compounds of porphyrins with divalent iron, carrying one or two axial ligands, with each hem containing four pyrrole rings (2). In total, this means that one hemoglobin molecule can actively bind and release up to four molecules of oxygen or 1.34 mL of dissolved gas (3). Consequently, the prosthetic center of hemoglobin molecules, represented by divalent iron, realizes the strong binding of four polypeptide chains, among which α1, α2, β1, and β2 are distinguished, as shown in Figure 1 (1). This variety of structural features of normal hemoglobin (or as it is often called hemoglobin A, HbA) leads to an overabundance of its potential forms.
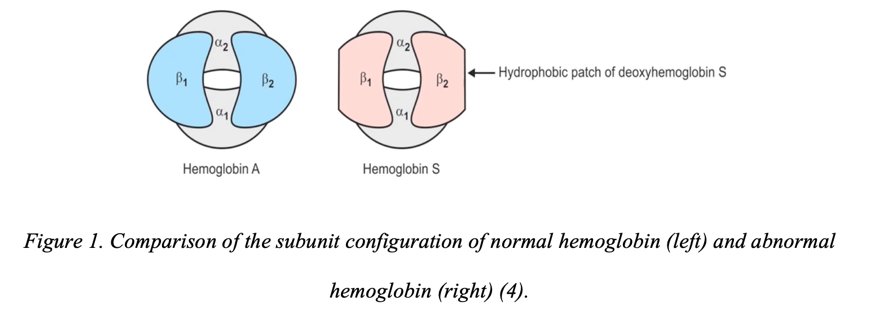
For example, if both beta-chains are replaced by delta-chains, this results in HbA2, which is a relatively rare but natural form of normal hemoprotein. Fetuses and newborn children also have HbF, the structure of which contains two gamma monomers in combination with alpha-chains. Moreover, the oxygen affinity of this protein is comparably higher than that of HbA, so much smaller volumes of blood are required for the adequate oxygenation of tissues. Normal forms of hemoglobin are limited to the proteins mentioned above, although it is fair to admit that there are a variety of abnormal variants. Among the most common are HbC, poorly oxygen-binding, mutagenic HbE, HbD, and, most interesting in the current study, HbS, as shown in Figure 1.
Genetic Factors of Abnormalization
It should be emphasized that the alpha- and beta-chains of normal hemoglobin are produced in different chromosomes. The alpha-chain genes are localized in the short arm of the 16th chromosome, and their total cluster is up to 30,000 base pairs. In contrast, the beta-chain genes are encoded by a sequence of 438 base pairs and are localized in the short arm of chromosome 11 at locus 15.4 (5). Consequently, the extended site of total hemoglobin synthesis increases the likelihood of deleterious mutations that can lead to abnormal protein structures, namely the formation of HbS instead of or in combination with HbA. When the results of biochemical and morphological analyses of hemoglobin structures show the presence of HbS in inpatient erythrocytes, this indicates the presence of a point mutation in the short arm of the eleventh chromosome. More specifically, the CTT triplet, which creates a matrix copy, GAA, on the informational RNA during translation, is localized at this position on the chromosome. This triplet is responsible for coding for glutamic acid, which forms the standard structure of normal hemoglobin (6). However, in a point mutation, the thymine at the second position is replaced by adenine, resulting in the triplet taking the form of CAT. As a result, a valine is generated at codon 6th of the first exon of the normal hemoglobin beta-chain gene instead of glutamic acid, making the total protein abnormal. Hence, the overall difference between HbA and HbS is the nature of the triplet within the gene that encodes the protein.
A point change in the nucleotide in the triplet entails many more mutagenic transformations than coding for a new amino acid. Abnormal hemoglobin forms as a result of a point single-nucleotide mutation have significant biochemical differences from the standard protein structure. Since the structure of the beta-chain has changed, the properties of the entire hemoglobin macromolecule also change: a sticky region is formed on its surface. The formation of the sticky site is caused by the replacement of glutamic acid with valine because, unlike glutamate, valine has a non-polar side chain (7). As a result, a non-polar residue emerges on the surface of the abnormal protein, which binds the individual HbS together into large agglomerated protein particles, as shown in Figure 2. Such clots have a gel-like consistency and are much less soluble (8). More specifically, if the concentration of abnormal protein exceeds 30 g/dL, HbS tends to aggregate into polymerase conglomerates. Such abnormal conglomerates can not only deform red blood cells, giving them a sickle shape but also provoke autohemolysis and tissue ischemia.
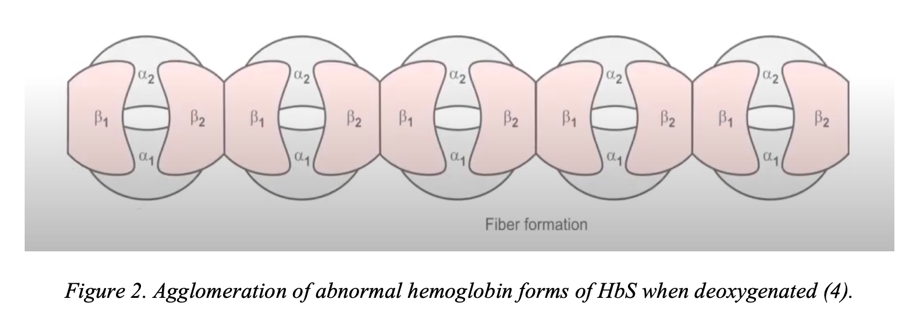
It happens that abnormal hemoglobins revert to their former, non-sickle shape during oxygenation, but this possibility is lost when the sickle effect accumulates. It is accurately known that 5% to 50% of all patient red blood cells have a rigid sickle-shaped fixation and cannot be rehabilitated over time (8). In other words, erythrocytes become incapable of taking a biconvex shape, and the patient progresses to a state of severe sickle cell anemia.
In addition, pathological changes initiated by biochemical transformations in the generation of abnormal protein are also observed at the ionic level. Preliminarily, it is worth recalling that sodium-potassium adenosine triphosphatase balances sodium and potassium ions by releasing the former into the external environment and accumulating the latter inside the cell (9). This maintains the resting potential of the cell and controls cellular volume. However, in sickle cell anemia, the situation is reversed: sodium ions are actively accumulated, and the intracellular potassium concentration rapidly decreases. This effect may be due to rapid cell dehydration and significant exposure to hypoxia, resulting in intracellular potassium release into the intercellular medium. Therefore, in sickle cell anemia, excessive concentrations of potassium ions can be detected in the serum of patients.
Disruption of adenosine triphosphatase also affects the active increase in the presence of calcium cations in the cytoplasm. This amount in sickle cell anemia is increased about fourfold compared to normal, resulting in increased erythrocyte membrane rigidity (9). In turn, this allows abnormal erythrocytes to retain their abnormal shape. It has been shown that a possible cause that initiates the appearance of sickle shape in red blood cells and the appearance of abnormal protein, in general, is increased adenosine signaling (10). More specifically, there is an increase in adenosine transmission through erythrocyte ADORA2B receptors, which leads to activation of the cellular AMPK protein kinase that controls cellular energy balance (Figure 3). In turn, this imbalance leads to an increase in the production of 2,3-BPG and, therefore, an increase in the polymerization processes of abnormal hemoglobins.
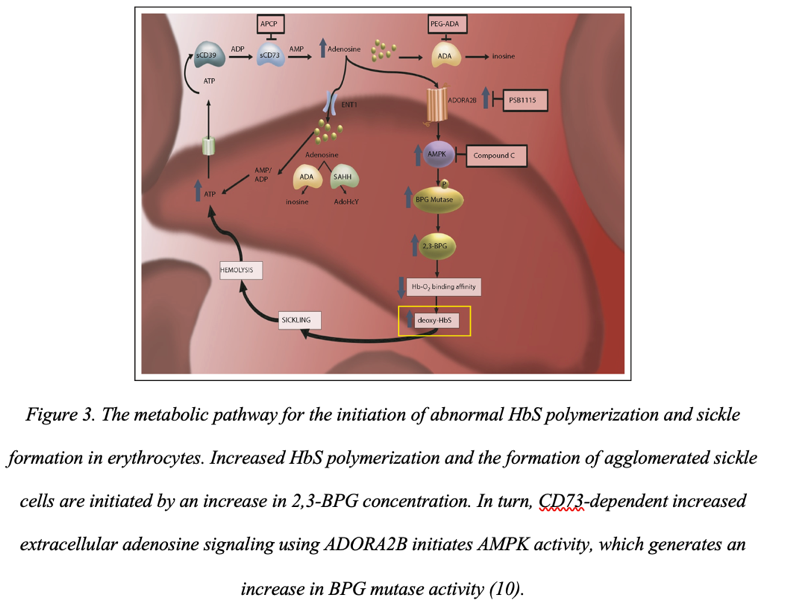
Another negative biochemical manifestation of sickle cell anemia is the increased adhesive capacity of abnormal red blood cells. In addition to the fact that HbS can stick together into conglomerates, abnormal red blood cells also have increased adhesiveness, causing them to attach to the surface of the endothelium (11). In turn, endothelial adhesion leads to endothelial dysfunction, which becomes the cause of crises. Adhesion is further initiated by hypoxia, as dissolved oxygen deficiency rapidly reduces nitric oxide production. As a result, the lack of nitric oxides in the blood leads to vasoconstriction, which only accelerates the initiation of thrombosis and tissue ischemia. Finally, bound sickle-shaped erythrocytes on the endothelium produce large amounts of VLA-4 antigen, which is responsible for increasing the adhesive properties of leukocytes (8). Thus, blood vessels from within tend to a state of complete dysfunction.
Consequently, due to spatial changes in the structure of normal hemoglobin, its abnormal confinement, HbS, occurs. The biochemical effect of this molecular change leads to the formation of a sticky site on the hemoglobin and a change in the shape of the erythrocytes from biconvex to sickle-shaped. This cell geometry prevents active oxygen binding, and consequently, the protein is no longer able to carry molecules of the vital element through the blood. The patient’s tissues are severely hypoxic, and since the disease is congenital, mental and physical developmental delays due to lack of oxygen are most likely.
Pathophysiological Manifestations
Sickle cell anemia is a hereditary disease that affects the human cardiovascular system. As has become apparent from the previous sections, in a family of carriers, the probability of getting their disease does not exceed 25%, but if a person gets it, their life is in grave danger. It should be noted that the clinical manifestation of this disease becomes evident by the first months of the life of the newborn when the amount of hemoglobin in their blood increases. With sickle cell anemia, the baby’s sickle cell count reaches the 90% mark by the fourth month, indicating the possibility of dangerous complications associated with hypoxia. In more detail, oxygen deficiency in the tissues of the central nerve fibers leads to inhibition of cell differentiation and, consequently, to developmental delay. The infant’s bone and cartilage formations also fail to develop because the lack of oxygen does not allow for forming a strong bone skeleton. Thus, a sick child often has acrocephaly, thickening of the frontal bones, lordosis, and kyphosis in uncharacteristic parts of the spine. In addition, it is evident that the degree of the disease closely correlates with the concentration of sickle cell erythrocytes in the blood: the higher their value, the more likely severe manifestations of sickle cell anemia.
In this regard, it is evident that this type of anemia requires careful medical care. Even seemingly insignificant factors can provoke complications. Stress conditions and even dehydration can lead to a hemolytic crisis, in which the patient’s red blood cells are destroyed (12). At the same time, about thirty percent of all patients report the development of autosplenectomy, during which spleen dysfunction is observed. In turn, this condition leads to a dramatic decrease in the patient’s immune status and, consequently, to more frequent cases of infectious diseases.
The Nature of Inheritance of the Mutation
Hereditary diseases can be transmitted both recessively and dominantly if the sex chromosomes are not involved. Sickle cell anemia is an autosomal recessive type of inheritance, which means that for the active form of the pathology to occur, the embryo must receive the recessive gene from both parents at once. In other words, the active form of the disease usually manifests itself if both copies of the genes from the parents have been damaged. As a result, such patients have an insufficient genetic and molecular basis for the synthesis of normal hemoglobin (or it is generated in trace amounts), and the entire basis of their hemoprotein is abnormal HbS. However, the autosomal recessive type of inheritance also determines that if both parents in a family were carriers of the defective gene, heterozygotes could also arise among their descendants. In this case, such children will generate both types of hemoglobin — normal and abnormal — but this does not mean that the offspring will be susceptible to sickle cell anemia (13). It seems evident that parents are interested in creating a healthy new generation whose children will not suffer from genetic diseases. For them, there is a clinical genetic screening procedure that can pinpoint gene damage in a couple and determine the likelihood of having unhealthy offspring.
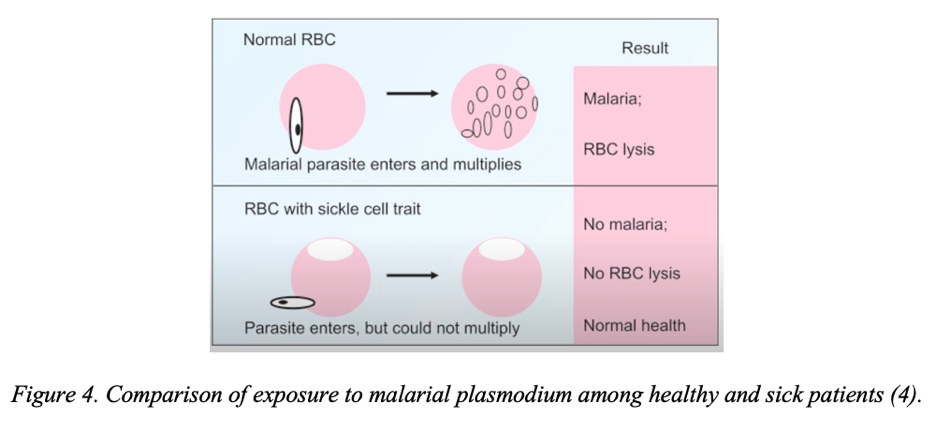
It is noteworthy that the leading share of those susceptible to the disease is the black population, especially in malaria-endemic regions. Recall that malaria is an acute infectious disease characteristic of southern countries in which a patient’s blood is infected with malarial plasmodium via an intermediate host in the form of a mosquito (14). In this case, the patient’s blood becomes infected with the pathogen, which gradually depresses it, which is manifested by fever, splenomegaly, hepatomegaly, and anemia. However, people with sickle cell anemia have a peculiar resistance to infection by malarial plasmodium. In particular, the abnormal shape of the protein prevents free fixation and entry of unicellulars inside the erythrocyte, making sickle-cell an escape from malaria, as shown in Figure 4 (15). Such claims are valid not only for homozygous patients but also for heterozygotes, in whom HbS is produced but in markedly reduced amounts. Thus, carriers demonstrate partial resistance to malaria but may be affected by it. In general, this character of resistance is a sufficient explanation of the reason for the wide prevalence of abnormal gene mutation in such regions. In the context of the statistical distribution, the greater susceptibility to genetic damage among the dark-skinned population is also confirmed. In fact, sickle cell anemia occurs in 1 in 365 black infants (16; 17). For whites, the rate is even lower, at 0.2% for infants.
Diagnosis and Treatment
Since the disease is hereditary, weakness and swelling in the extremities caused by pathological conditions in the peripheral bones are the first signals of its manifestation. An exact diagnosis can also be made by blood tests and a detailed morphological examination of the hemoglobin structures later on. In addition, because of the heredity of the disease, the therapist will need a family history of the disease since the recessive nature of sickle cell anemia implies a manifestation of the disease in the ancestors. However, the best and earliest result can be obtained with a prenatal diagnosis of the disease. Specifically, DNA sampling from the chorion can be performed as early as 8-12 weeks of pregnancy, so the physician will have time to prepare the family (18). The primary treatment is aimed at preventing the patient from developing infections, so weekly doses of penicillin may be used (19). Another vector of therapy is to stimulate hematopoiesis processes so that more red blood cells are produced in the blood. For this purpose, hematologists often recommend folic acid, which is a natural enzyme (20). However, clinical developments to treat the disease do not stop there. Studies show that biocompatible blood transfusions from a healthy patient, the use of the antitumor hydroxycarbamide, and even transplantation of the hematopoietic stem cell system may prove to be useful tools for treating the patient (21). On the other hand, recognizing blood transfusion as a very resource-intensive procedure, the authors have proposed the use of a natural ribonucleoside diphosphate reductase inhibitor that is a target for antitumor drugs (22). More specifically, the enzyme catalyzes the reduction of ribonucleotides to deoxyribonucleotides and thus regulates the activity of the limiting reaction in DNA replication. Thus, with timely diagnosis, treatment of sickle cell anemia is possible and actively practiced even before birth. Even for an adult patient, there are effective therapeutic options.
Conclusion
In conclusion, among the severe hereditary diseases, special attention should be paid to sickle cell anemia, which negatively affects the patient’s circulatory system. With mutations in the eleventh chromosome, a point nucleotide substitution occurs, resulting in the replacement of glutamic acid in the protein with valine. The change in the beta chain entails abnormalization of total hemoglobin: it has increased adhesive properties. HbS tends to combine into polymers and attach to the endothelium, resulting in erythrocyte dysfunction. There is a treatment, but for maximum efficacy, it is necessary to identify the possibility of disease development as early as possible.
References
AMBOSS. (2021). Erythrocyte morphology and hemoglobin. AMBOSS. Web.
Kleingardner, J. G., Levin, B. D., Zoppellaro, G., Andersson, K. K., Elliott, S. J., & Bren, K. L. (2018). Influence of heme c attachment on heme conformation and potential. Journal of Biological Inorganic Chemistry, 23(7), 1073-1083.
Rizvi, A., Macedo, P., Babawale, L., Tighe, H. C., Hughes, J. M. B., Jackson, J. E., & Shovlin,
C. L. (2017). Hemoglobin is a vital determinant of arterial oxygen content in hypoxemic patients with pulmonary arteriovenous malformations. Annals of the American Thoracic Society, 14(6), 903-911.
Amit. (2020). Sickle cell anemia biochemistry || sickle cell anemia [Video]. Web.
NCBI. (2021). Human hemoglobin A beta chain. NCBI. Web.
NIH. (2020). About sickle cell disease. NIH. Web.
Pratapa, S. K., Acharya, S., Gupte, Y., & Shukla, S. (2020). Acute B virus hepatitis with fulminant hepatic failure precipitating a crisis in sickle cell disease [PDF document]. Web.
Maakaron, J. E. (2021). Sickle cell anemia. Medscape. Web.
Waugh, D. T. (2019). Fluoride exposure induces inhibition of sodium and potassium-activated adenosine triphosphatase (Na+, K+-ATPase) enzyme activity: Molecular mechanisms and implications for public health. International journal of environmental research and public health, 16(8), 1427-1441.
Adebiyi, M. G., Manalo, J. M., & Xia, Y. (2019). Metabolomic and molecular insights into sickle cell disease and innovative therapies. Blood Advances, 3(8), 1347-1355.
Zhang, J. (2019). Regulation of sickle cell disease-specific erythrocyte adhesion [PDF document]. Web.
Brugnara, C. (2018). Sickle cell dehydration: Pathophysiology and therapeutic applications. Clinical Hemorheology and Microcirculation, 68(2-3), 187-204.
Sickle cell anemia: A look at the connection between DNA and phenotype [PDF document]. Web.
White, N. J. (2018). Anemia and malaria. Malaria Journal, 17(1), 1-17.
Uyoga, S., Macharia, A. W., Ndila, C. M., Nyutu, G., Shebe, M., Awuondo, K. O.,… & Williams, T. N. (2019). The indirect health effects of malaria are estimated from the health advantages of the sickle cell trait. Nature Communications, 10(1), 1-7.
CDC. (2020). Data & statistics on sickle cell disease. CDC. Web.
Ojodu, J., Hulihan, M. M., Pope, S. N., & Grant, A. M. (2014). Incidence of sickle cell trait—the United States, 2010. Morbidity and Mortality Weekly Report, 63(49), 1155-1167.
Short, L., Baah, W., & Oteng-Ntim, E. (2018). A systematic review and meta-analysis of non-invasive prenatal diagnosis (NIPD) of sickle cell disease (SCD). Global Journal of Hematology and Blood Transfusion, 5(1), 1-11.
Fischer, P. R. (2018). Pneumococcus, sickle cell disease, vaccination, and penicillin. Infectious Disease Alert, 37(5), 1-13.
Williams, B. A., McCartney, H., Adams, E., Devlin, A. M., Singer, J., Vercauteren, S.,… & Karakochuk, C. D. (2020). Folic acid supplementation in children with sickle cell disease: Study protocol for a double-blind randomized cross-over trial. Trials, 21(1), 1-11.
Kato, G. J., Piel, F. B., Reid, C. D., Gaston, M. H., Ohene-Frempong, K., Krishnamurti, L.,… & Vichinsky, E. P. (2018). Sickle cell disease. Nature Reviews Disease Primers, 4(1), 1-22.
Neumayr, L. D., Hoppe, C. C., & Brown, C. (2019). Sickle cell disease: current treatment and emerging therapies. Am J Manag Care, 25(18), 335-343.